9. Commands for Hi-C and Micro-C processing#
CustardPy internally executes Juicer and juicertools. See the original website for the full description of each command.
9.1. Juicer analysis#
9.1.1. custardpy_juicer#
custardpy_juicer is an end-to-end pipeline for Juicer analysis.
It executes juicer_map.sh, juicer_pigz.sh, plot_distance_count.sh,
juicer_callTAD.sh, call_HiCCUPS.sh, makeMatrix_intra.sh, makeEigen.sh, and makeInsulationScore.sh.
The results are stored in CustardPyResults/Juicer_$build/$prefix/.
custardpy_juicer [Options] <fastqdir> <label>
<fastqdir>: directory that contains input fastq files (e.g., "fastq/sample1")
<label>: label of the input (e.g., "sample1")
Options:
-i index : bwa index
-g genometable : genome table file (describing the chromosome length)
-e enzyme : enzyme (HindIII|MboI|DpnII|Sau3AI|Arima, default: HindIII)
-b build : genome build (hg19|hg38|mm10|mm39|rn7|galGal5|galGal6|ce10|ce11|danRer11|dm6|xenLae2|sacCer3|T2T)
-z [_|_R]: if the filename of fastqs is *_[1|2].fastq, supply "_". if *_[R1|R2].fastq, choose "_R". (default: "_")
-o outputdir : output directory (default: 'CustardPyResults')
-n [NONE|VC|VC_SQRT|KR|SCALE] : normalization type (default: SCALE)
-a <refFlat>: gene annotation file
-r resolutions : resolutions for 1D metrics calculation (default: "25000 50000 100000", should be quoted and separated by spaces)
-p ncore: number of CPUs (default: 32)
-m tmpdir: tempdir
-L: Allocate larger memory ("-Xms1024m -Xmx655360m", default: "-Xms512m -Xmx65536m", for deep-sequenced samples; e.g., Rao 2014)
Example:
custardpy_juicer -i bwaindex/hg38 -g genometable.hg38.txt -b hg38 -e HindIII -z _R -a refFlat.hg38.txt fastq/Hap1-A Hap1-A
9.1.2. juicer_map.sh#
juicer_map.sh map FASTQ files using BWA and generates .hic file using juicertools.
The input FASTQ files can be gzipped (.fastq.gz).
The results including the .hic file is outputted in $odir.
The BWA index of the reference genome is necessary.
juicer_map.sh [options] <fastqdir> <odir> <build> <gt> <bwaindex> <enzyme> <fastq_post>
<fastqdir>: directory that contains input fastq files (e.g., "fastq/sample1")
<odir>: output directory (e.g., "JuicerResults/sample1")
<build>: genome build (e.g., hg38)
<gt>: genome table
<bwaindex>: index file of BWA
<enzyme>: enzyme (e.g., HindIII, MboI)
<fastq_post [_|_R]>: if the filename of fastqs is *_[1|2].fastq, supply "_". if *_[R1|R2].fastq, choose "_R".
Options:
-p ncore: number of CPUs (default: 32)
-m tmpdir: tempdir
-L: Allocate larger memory ("-Xms1024m -Xmx655360m", default: "-Xms512m -Xmx65536m", for deep-sequenced samples; e.g., Rao 2014)
Example:
juicer_map.sh $(pwd)/fastq/Hap1-A/ $(pwd)/JuicerResults/Hap1-A hg38 genometable.hg38.txt bwaindex/hg38 HindIII _R
Note
The input FASTQ files of each sample should be stored in the separated directory.
For example, if there are three Hi-C samples (sample1, sample2, and sample3), the fastq files should be in fastq/sample1/, fastq/sample2/, and fastq/sample3/.
Output
merged_nodups.txt … mapped fragments
inter.hic … .hic file (without quality filtering)
inter.txt … stats for inter.hic
inter_30.hic … .hic file (Q>=30)
inter_30.txt … stats for inter_30.hic
We recommend using inter_30.hic for the downstream analysis.
9.1.3. juicer_pigz.sh#
Since the output files of Juicer are quite large, CustardPy provides a script juicer_pigz.sh that compresses the intermediate files.
This command is optional while custardpy_juicer implements it.
juicer_pigz.sh <odir>
<odir> output directory of juicer_map.sh (e.g., "JuicerResults/sample1")
Note that some commands provided in Juicer use the intermediate files (e.g, mega.sh).
Because these commands do not accept the compressed format, use juicer_unpigz.sh that uncompresses the compressed files.
juicer_unpigz.sh <odir>
<odir> output directory of juicer_map.sh (e.g., "JuicerResults/sample1")
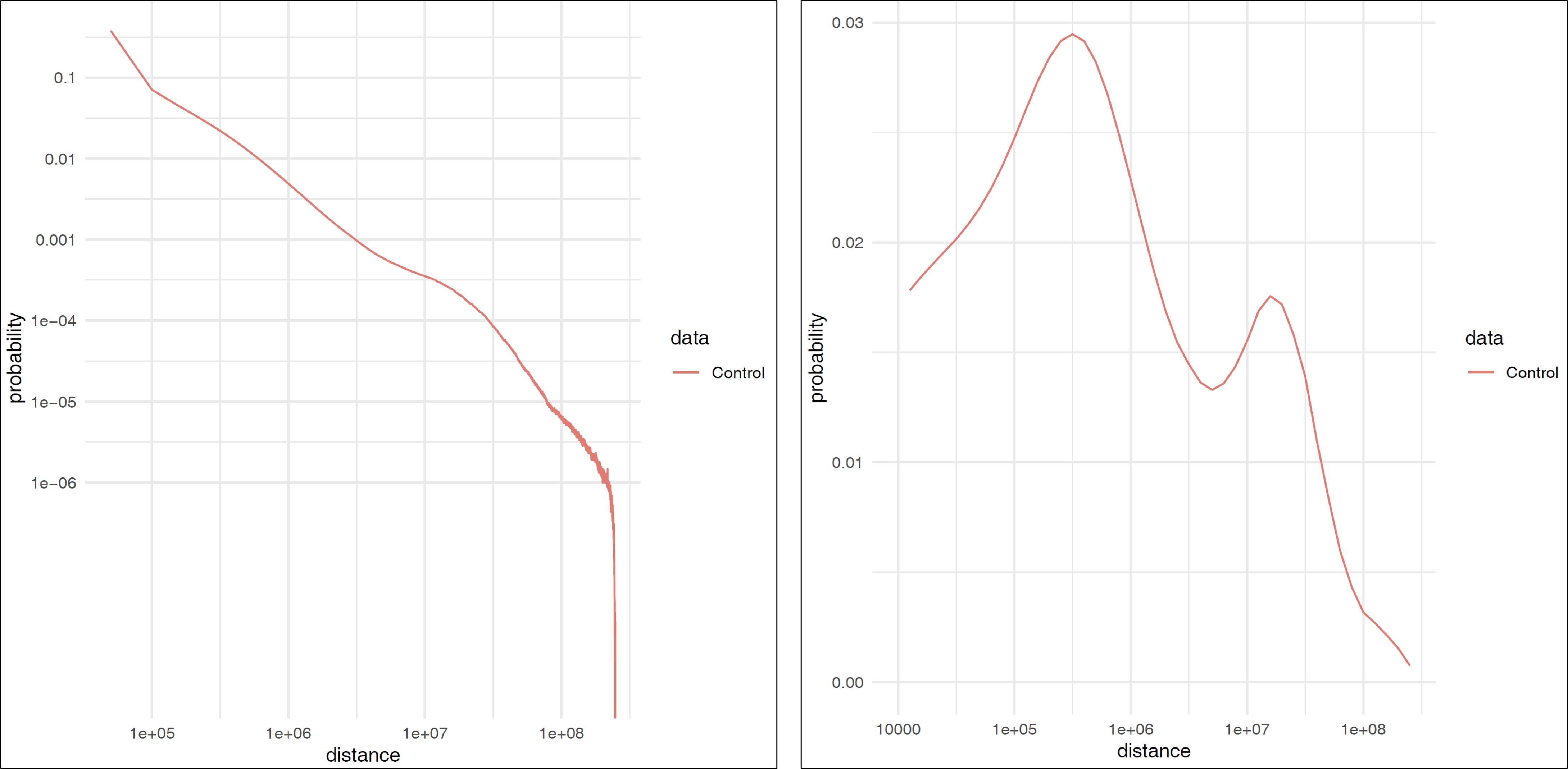
9.1.4. plot_distance_count.sh#
plot_distance_count.sh calculates the fragment distance and generates a figure (.pdf).
The result is outputted in distance/ directory.
plot_distance_count.sh <label> <odir>
<label>: title of the figure
<odir> output directory of juicer_map.sh (e.g., "JuicerResults/sample1")
Output
distance_vs_count.10kb.MAPQ30.pdf … figure of distance plot
distance_vs_count.10kb.MAPQ30.txt … values for the plot
distance_vs_count.10kb.MAPQ30.log.pdf … figure of distance plot (log scale)
distance_vs_count.10kb.MAPQ30.log.txt … values for the plot (log scale)
9.1.5. custardpy_process_hic#
custardpy_process_hic takes a .hic file as input and executes juicer_callTAD.sh, call_HiCCUPS.sh, makeMatrix_intra.sh, makeEigen.sh, and makeInsulationScore.sh.
custardpy_process_hic [Options] <hicfile> <odir>
<hicfile>: .hic file generated by Juicer
<odir> : output directory
Options:
-g genometable : genome table file (describing the chromosome length)
-n [NONE|VC|VC_SQRT|KR|SCALE] : normalization type (default: SCALE)
-a <refFlat>: gene annotation file
-r resolutions : resolutions for 1D metrics calculation (default: "25000 50000 100000", should be quoted and separated by spaces)
-p ncore: number of CPUs (default: 32)
-o: Use older version of juicer_tools.jar for old .hic files (juicer_tools.1.9.9_jcuda.0.8.jar, default: juicer_tools.1.22.01.jar)
Example:
custardpy_process_hic -g genometable.hg38.txt -a refFlat.hg38.txt Hap1-A/inter_30.hic Hap1-A
Note
custardpy_process_hic fails when .hic files created by the old version of juice_tools.jar are applied because they do not contain the string “chr” in the chromosome name. In such a case, supply -o option in custardpy_process_hic, which uses the old version of juicer_tools.jar (juicer_tools.1.9.9_jcuda.0.8.jar).
9.1.6. makeMatrix_intra.sh#
makeMatrix_intra.sh takes a .hic file as input and generates the matrices of intra-chromosomal interactions for all chromosomes. The chromosome Y and M are omitted.
makeMatrix_intra.sh <norm> <odir> <hic> <resolution> <gt>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<hic>: .hic file
<resolution>: resolution of the matrix
<gt>: genome table
Options:
-l: output contact matrix as a list (default: dense matrix)
-o: Use older version of juicer_tools.jar for old .hic files (juicer_tools.1.9.9_jcuda.0.8.jar, default: juicer_tools.1.22.01.jar)
The resulting observed/oe matrices are output in <odir>/Matrix/intrachromosomal/<resolution>/.
9.1.7. makeMatrix_inter.sh#
makeMatrix_inter.sh generates the inter-chromosomal interactions matrix for a specified chromsome pair.
makeMatrix_inter.sh [-l] <norm> <odir> <hic> <resolution> <chr1> <chr2>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<hic>: .hic file
<resolution>: resolution of the matrix
<chr1, chr2>: two input chromosomes
Options:
-l: output contact matrix as a list (default: dense matrix)
-o: Use older version of juicer_tools.jar for old .hic files (juicer_tools.1.9.9_jcuda.0.8.jar, default: juicer_tools.1.22.01.jar)
The resulting observed/oe matrices are output in <odir>/Matrix/interchromosomal/<resolution>/<chr1>-<chr2>.
9.1.8. makeEigen.sh#
makeEigen.sh generates eigenvector file (compartment PC1) from a .hic file using HiC1Dmetrics.
The sign (+-) of the value indicating A/B compartments is adjusted by the number of genes.
makeEigen.sh [options] <norm> <odir> <hic> <resolution> <genometable> <refFlat>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<hic>: .hic file
<resolution>: resolution of matrix
<genometable>: genometable file
<refFlat>: gene annotation file (refFlat format)
Options:
-p <int>: the number of CPUs (default: 6)
9.1.9. juicer_callTAD.sh#
juicer_callTAD.sh calls TADs from a .hic file using Juicer ArrowHead.
juicer_callTAD.sh [options] <norm> <odir> <hic> <gt>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<hic>: .hic file
<gt>: genome table
Options:
-r resolutions: the resolutions for ArrowHead (default: "10000 25000 50000", should be quoted and separated by spaces)
-p ncore: number of CPUs (default: 24)
-o: Use older version of juicer_tools.jar for old .hic files (juicer_tools.1.9.9_jcuda.0.8.jar, default: juicer_tools.1.22.01.jar)
- Output:
\*_blocks.bedpe… TAD regions (BEDPE format, default output of Juicer ArrowHead)\*_blocks.bed… TAD regions (BED format file converted from\*_blocks.bedpe)\*_blocks.merged.bed… Non-overlapped TAD list (overlapped TAD are merged bybedtools merge)\*_blocks.boundaries.bed… TAD boundaries (“inside” window of called TADs, including boundaries of nested TADs)\*_blocks.TADcoverage.bed… Number of TADs that cover the genomic positions (for nested TAD analysis)\*_blocks.TADregions.bed… List of intra-TAD regions (inside of TAD boundaries)\*_blocks.nonTADregions.bed… List of regions that are not covered by any TADs
Note
Because Juicer ArrowHead allows “nested TADs” and “non-TAD regions”, not all genomic regions are included in TADs, and some amount of TAD boundaries may be included in a larger TADs. Make sure that the files you are using meet the criteria of your assumption.
9.1.10. makeInsulationScore.sh#
makeInsulationScore.sh takes the observed matrices files generated by makeMatrix_intra.sh as input and calculates the insulation score for all chromosomes. The chromosome Y and M are omitted.
The <odir> directory should be the same with that is specified in makeMatrix_intra.sh.
makeInsulationScore.sh <norm> <odir> <resolution> <gt>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<resolution>: resolution of the matrix
<gt>: genome table
The results are output in <odir>/InsulationScore/<norm>/<resolution>/.
9.1.11. call_HiCCUPS.sh (GPU required)#
call_HiCCUPS.sh calls loops using Juicer HiCCUPS.
Supply --gpus all for Docker and --nv option for Singularity to activate GPU as follows:
singularity exec --nv custardpy_juicer.sif call_HiCCUPS.sh
docker run --rm -it --gpus all rnakato/custardpy call_HiCCUPS.sh
call_HiCCUPS.sh <norm> <odir> <hic>
<norm>: normalization type (NONE|VC|VC_SQRT|KR|SCALE)
<odir>: output directory (e.g., "JuicerResults/sample1")
<hic>: .hic file
Options:
-r resolutions: the resolutions (default: "5000,10000,25000", should be quoted and separated by comma)
-o: Use older version of juicer_tools.jar for old .hic files (juicer_tools.1.9.9_jcuda.0.8.jar, default: juicer_tools.1.22.01.jar)
Output
merged_loops.simple.bedpe … loop file
9.1.12. call_MotifFinder.sh#
If you have peak files of cohesin and CTCF, you can use MotifFinder by call_MotifFinder.sh:
call_MotifFinder.sh <build> <motifdir> <loop>
<build>: genome build
<motifdir>: the directory that contains the BED files
<loop>: loop file (.bedpe) obtained by HiCCUPS
If the <build> is (hg19|hg38|mm9|mm10), this command automatically supplies FIMO motifs provided by Juicer.
Output
merged_loops_with_motifs.bedpe
See MotifFinder manual for more information.
Note
Because an error occurs in the latest version of juicertools, CustardPy uses juicertools version 1.9.9 for MotifFinder.
9.2. Cooler analysis#
9.2.1. custardpy_cooler#
custardpy_cooler uses Cooler to generates .cool and .hic files from FASTQ files.
The input FASTQ files can be gzipped (.fastq.gz).
BWA and chromap can be used for mapping reads (use -M option).
The results are stored in CustardPyResults/Cooler_$build/$prefix/.
The index file of BWA or chromap (-i <index>) and the fasta file of the reference genome (-f <genome>) are required.
custardpy_cooler [options] -i <index> -g <gt> -f <genome> <fastqdir> <odir>
fastqdir: Directory that contains input fastq files (e.g., "fastq/")
odir: Name of output directory
Options:
-M mapper : mapping tool for mapping reads [bwa|chromap] (default: bwa)
bwa: use BWA for mapping
chromap: use Chromap for mapping
-i index : index for BWA or chromap
-g genometable : genome table file (describing the chromosome length)
-f genome file : fasta file of the reference genome (original data of the index files)
-w : apply restriction site filtering (default: not applied)
-e enzyme : enzyme (HindIII|MboI|DpnII|Sau3AI|None, default: HindIII)
-b build : genome build (hg19|hg38|mm10|mm39|rn7|galGal6|ce11|danRer11|dm6|xenLae2|sacCer3|S.pombe|HVAEP)
-o outputdir : output directory (default: 'CustardPyResults')
-q qvalue : threshold of mapped fragments (default: 30, for '--min-mapq' of pairtools parse)
-p ncore : number of CPUs (default: 4)
-z [_|_R]: if the filename of fastqs is *_[1|2].fastq, supply "_". if *_[R1|R2].fastq, choose "_R". (default: "_")
-m max_distance : 8.4 for human, 8.2 for mouse (for pairsqc.py, default: 8.4)
-n binsize_min : binsize_min (for cooler cload pairix, default: 5000)
-r binsize_multi : binsize_multi (for multirescool, default: '5000,10000,25000,50000,100000,500000,1000000,2500000,5000000,10000000')
Output
cool/ … directory for .cool files
hic/ … directory for the .hic file
log/ … log files of mapping
mapfile/ … directory of mapping file
pairs/ … .pairs file generated by pairtools
qc_report/ … directory for statistics and QC files
9.2.2. calculate_compartment_strength#
calculate_compartment_strength calculates the compartment strength from Hi-C data using GENOVA.
calculate_compartment_strength <coolfile> <sample name>
coolfile: Input Hi-C data (.cool format)
sample name: Name of the sample (also used for the output file name)
<Example>
calculate_compartment_strength Cooler_results/Control/coolfile/Control.25000.cool Control
The output file is [sample_name].GENOVA_compartment_score.txt containing the compartment strength, which is an average score for the chromosomes.
9.3. Utility tools#
9.3.1. run_3DChromatin_ReplicateQC.sh#
Since it is written in Python2.7, we use a virtual environment in the CustardPy docker image.
run_3DChromatin_ReplicateQC.sh`` is a script to run it from the default command line.
Replace 3DChromatin_ReplicateQC with run_3DChromatin_ReplicateQC.sh in the command line:
$ run_3DChromatin_ReplicateQC.sh -h
usage: 3DChromatin_ReplicateQC [-h]
{run_all,preprocess,qc,concordance,summary,cleanup}
...
positional arguments:
{run_all,preprocess,qc,concordance,summary,cleanup}
run_all Run all steps in the reproducibility/QC analysis with
this single command
preprocess (step 1) split files by chromosome
qc (step 2.a) compute QC per sample
concordance (step 2.b) compute reproducibility of replicate pairs
summary (step 3) create html report of the results
cleanup (step 4) clean up files
optional arguments:
-h, --help show this help message and exit
9.3.2. distance_vs_count.Juicer#
Count the genomic distance of read pairs in the input file (supposing align/merged_nodups.txt.gz in Juicer outputs)
The output file can be used for the distance plot with plot_distance_count.R.
distance_vs_count.Juicer <file> <winsize> <MAPQ>
<file>: Input file (merged_nodups.txt.gz)
<winsize>: window size (default: 10000)
<MAPQ>: MAPQ threshold (default: 30)
Example:
distance_vs_count.Juicer align/merged_nodups.txt.gz 50000 30
9.3.3. distance_vs_count.Juicer.log#
Count the genomic distance of read pairs in the input file with the log-scale bins.
The output file can be used for the distance plot with plot_distance_count.log.R.
Usage: distance_vs_count.Juicer.log <file> <MAPQ>
<file>: Input file (merged_nodups.txt)
<MAPQ>: MAPQ threshold (default: 30)
Example:
distance_vs_count.Juicer.log align/merged_nodups.txt.gz 30
9.3.4. convert_JuicerDump_to_dense.py#
Convert interaction frequency file dumped by Juicertools to dense (two-dimensional) matrix.
convert_JuicerDump_to_dense.py <inputfile> <outputfile> <genometable> <chr> <resolution> [--help]
Example:
convert_JuicerDump_to_dense.py \
Matrix.observed.VC_SQRT.chrX.txt \
Matrix.observed.VC_SQRT.chrX.matrix.gz \
genome_table.txt \
chrX \
25000
9.4. Loop analysis#
9.4.1. Fit-Hi-C#
Fit-Hi-C is a tool for loop calling. It takes the pairs file generated by Cooler as input and outputs the significant interactions.
Note that the original Fit-Hi-C takes the .pair file for input, but we modified it to use the .cool file instead.
cell=Control
gt=genometable.hg38.txt
resolution=25000
odir=CustardPyResults/Cooler_hg38/$cell
cool=$odir/cool/contact.bwa.q30.mcool
odir_fithic=$odir/loops/fithic/$resolution
run_fithic.sh -g $gt $cool $odir_fithic $cell $resolution
The generated loop file after merging is stored in
$odir/loops/fithic/$resolution/<label>.merged.gz.The
$odir/loops/fithic/$resolution/<label>.spline_pass1.res$resolution.significances.txt.gzfile is an intermediate file containing all candidate interactions including non-significant ones.The HTML report is stored in
$odir/loops/fithic/$resolution/<label>.results.html.The log file of the
fithic.pyandmerge-filter.shis stored in$odir/loops/fithic/$resolution/<label>.fithic.logand$odir/loops/fithic/$resolution/<label>.merged.log.
9.5. Stripe analysis#
9.5.1. Stripenn#
Stripenn is a tool for stripe calling and is installed in the virtual environment.
You can launch it using the run_env.sh command.
# For Docker
docker run --rm -it -u root rnakato/custardpy run_env.sh stripenn stripenn
# For Apptainer
apptainer exec custardpy.sif run_env.sh stripenn stripenn
9.6. HiChIP analysis#
9.6.1. FitHiChIP#
CustardPy includes FitHiChIP to process HiChIP data.
It uses HiC-Pro, which is installed in the hic-pro virtual environment.
You can use FitHiChIP with the following command:
run_env.sh hic-pro FitHiChIP_HiCPro.sh -C <configfile>
run_env.sh is a script to activate the hic-pro environment and execute the commands of FitHiChIP.
If you want to use any commands in the hic-pro environment, use the run_env.sh script:
run_env.sh hic-pro <command>